|
Electronic energy level alignment at metal-molecule interfaces with a GW approach
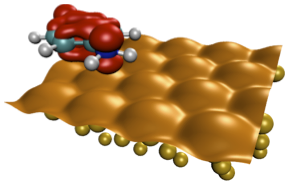
I. Tamblyn, P. Darancet, S.Y. Quek, S.A. Bonev, and J.B. Neaton Physical Review B, 84, 201402(R) (2011) This work employs density functional theory and many-body perturbation theory within the GW approximation to examine electronic energy level alignment at the interface between benzene diamine molecules and a gold surface. Standard density functional calculations are found to underestimate the molecular resonance energy by up to 0.8 eV compared to photoemission data, with the error arising from two sources: an underestimation of the molecule's gas-phase ionization energy by 0.7 eV and an overestimation of the gold work function by 0.2 eV. By refining the self-energy calculations to account for these deviations, the authors demonstrate quantitative agreement with experiment, providing a systematic framework for accurately predicting electronic structure at metal-molecule interfaces. |