|
|
Machine Learning for Advanced Materials
| Using a combination of theoretical tools (multi-scale modelling), our group is exploring and designing advanced materials. Our goal is to make materials that are, in the words of Mervin Kelly, "better, cheaper, or both".
The long-term objective of my research is to teach a computer chemical and physical intuition. A trained artificial intelligence (AI) should be able to make informed decisions about how to solve problems in chemistry, physics, and nanoscience without human intervention. Prediction and understanding are not synonyms; pushing modern AI methods to achieve chemical and physical understanding is central to my research focus. Although I do not expect to reproduce a scientist in silico within the next 5 years, I do believe that we are approaching an era where machines will learn chemical design rules based on a combination of theoretical models and experience, both simulated and real. The central question of my research program is how we can enhance discovery and scientific understanding through the use of modern AI and deep learning. We categorize our efforts into two sub-areas: 1) development of an accurate, scalable, and interpretable approach to the electronic structure problem - Accelerated Electronic Structure (AES). AES will make predictive calculations faster, cheaper, and more scalable while retaining the key element of human interpretability. More recently, we have also begun work on 2) AI controlled molecular self-assembly and growth (i.e. Self-Driving Chemistry, SDC) for solving the material inverse design problem. Recent Progress: In the same way that a computer can learn to identify different animal species, fruit, or people in a photograph by observing many training examples, we have shown that deep neural networks can accurately learn the structure-to-property mapping at the nanoscale. So far, our group has shown that deep neural networks have the ability to replace both classical [33, 41, 45] and quantum mechanical operators [32, 34, 36, 37, 40, 44, 46, S_HHG]. In comparison to other machine learning methods, we demonstrated that convolutional deep neural networks prevailed as the most accurate and best parallel-scaling method for all but the most simple physical problems [29]. We demonstrated the ability of deep neural networks to learn both the mapping from spin-configuration-to-energy (for the case of the ferromagnetic Ising model and screened Coulomb interaction) as well as magnetization. Using both machine-learned operators simultaneously, we are able to reproduce the temperature-induced order-to-disorder phase transition the Ising model is famous for (at the correct critical temperature, Tc) [33]. Extending this work with unsupervised learning [45], we were able to accurately extrapolate across thermodynamic conditions and phase boundaries. We also demonstrated that generative adversarial networks can be used to efficiently reproduce experimental observations of optical quantum lattices [S_RUGAN] and can be used to generate accurate mesoscale surfaces and nanoscale chemical motifs [44]. For a confined electron, our deep neural networks successfully learned the energy of the ground state, first excited state, and its kinetic energy [32]. We then used a similar approach to map the structure of a 2D material to energies computed within the density functional theory [34], where we trained the system to learn the mapping between a pseudopotential and total energy. All of this was accomplished via "featureless" deep learning, meaning we presented the network with raw, spatial data, without any form of manual feature selection. Overcoming a scaling limitation with traditional deep neural networks, we developed a new architecture and training protocol which naturally enforces extensivity onto the system [36]. Our new extensive deep neural networks (EDNN) are able to learn the locality length-scales of operators and look extremely promising for providing efficient alternative electronic structure methods to density functional theory (DFT) [34]. DFT is the most widely used electronic structure method in the world and is used throughout chemistry, physics, and material science both to interpret and make predictions for experiments. Our new EDNN are an O(N) method - so far we have seen that extreme predictive speedups are possible (over 1 million times faster than DFT) without any loss of accuracy. They overcome the normal O(N^3) scaling issue with Kohn-Sham DFT because they naturally learn the correlation length-scale of operators, and can decompose a material into sub-regions based on this learned scale. Applying EDNN to density functional theory resulted in the best performance and scaling ever demonstrated, enabling rapid searches through material space [40]. We have also experimented with deep convolutional inverse graphics networks and found that they are able to accurately predict the time-dependent dipole moment of excited-state molecules; they are a stand-in replacement for numerical solutions to the time-dependent Schrodinger equation [S_HHG]. In pursuit of enabling Self-Driving Chemistry, our recent work has consisted of investigating deep reinforcement as a control scheme for thermodynamic processes and rare events [41]. Here we demonstrated that the same ideas which can produce an expert AI video-game player can be applied to controlling dynamical systems. Using this insight, we were able to demonstrate improvements to classical optimization algorithms for a broad class of problems (quadratic unconstrained binary optimization, see [43]) relevant to adiabatic quantum computing. A crucial component of building self-driving laboratories will be the development of interpretable and reliable methods which can guide discovery. We are making significant progress in this direction [S_DARWIN, S_TNNR]. It is not enough just to develop a "black box"; we require methods which are physically motivated and can provide scientists with intuition and guidance [47]. Most recently, our efforts have been focused on understanding and applying techniques from reinforcement learning to physics and chemistry. These include heat engines, chemical reactions, and non-equilibrium self-assembly of nanostructure materials (topics I have previously published on, see here). To do this, we are developing deep learning methods which can rapidly approximate the properties of materials based on first principles training data. Ultimately, our goal is to develop a model which is transferrable, generalizable, reliable, and scalable. Transferability means that the network is able to accurately predict properties for structures which it has not observed during training. Generalizability means that the approach can be used for arbitrary observables, and that it is not sensitive to the details of the physical system to which it applies. For example, we would like to be able to work with fluids, continuum models, and atomistic systems using the same basic methodologies, model architectures, and training protocols. By reliable, we mean that the model is able to provide accurate estimates for a property, and, perhaps more importantly, provide the user with a warning or signal when it is uncertain. In our experience, neural networks are much better at interpolating than extrapolating. We have found that whenever a model has difficulty with a particular configuration (i.e. the error is high), the configuration is far away from the training set (in weight space). In practice, one obviously would not have access to the "ground truth" value, therefore a reliable model should be able to signal not only its prediction, but also a measure of confidence. The final property, scalability, relates to the extent to which the computational cost of evaluating the model can be distributed (e.g. across nodes in a cluster or multiple GPU). Our latest approaches are generalizable, scalable, and transferrable. Their reliability can be ensured through a GAN-like training procedure
Recent articles
|
|
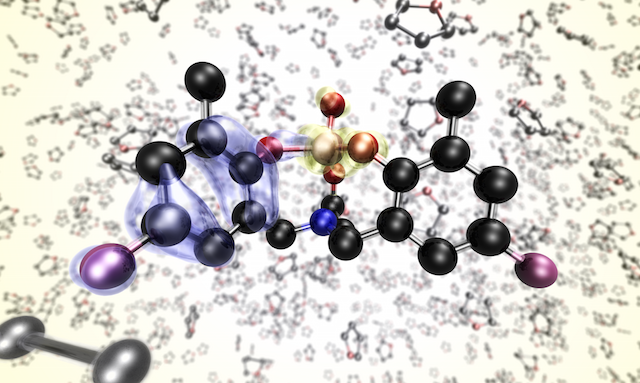
Ab initio electrolysis
Given the ubiquity of electrochemistry as an analytical and industrial tool, the importance of developing a fully first principles description of this system cannot be overstated.
At the same time, the complexity of the problem poses a significant obstacle to the development of an accurate atomistic description.
Obtaining the correct physical picture will require the union of several areas of theory.
A complete treatment will require an accurate description of the electrode-solution interface, both in terms of local geometry and electronic structure.
Methodological development will be important in this field.
Recent articles |
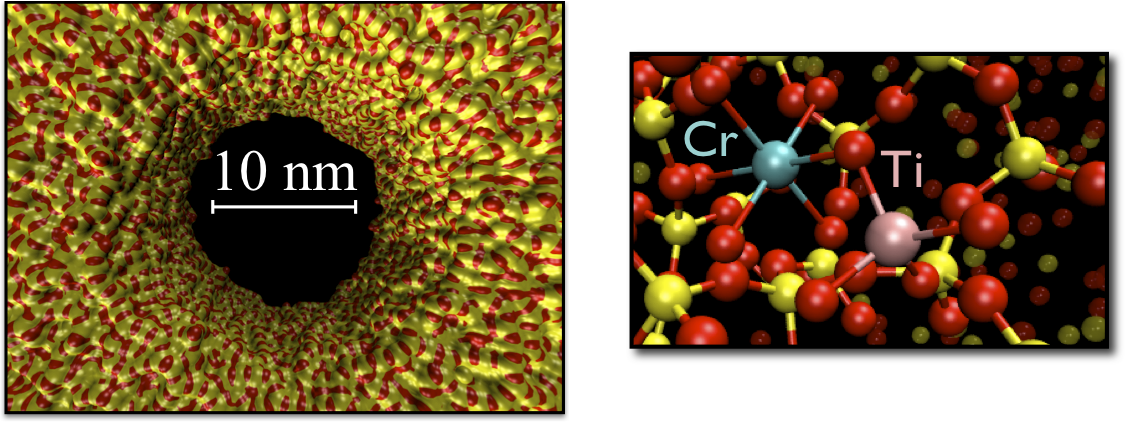
Energy harvesting materials
|
Light from the sun provides an unlimited supply of energy. Our challenge is to harness this energy in an efficient and scalable manner. Using metal-to-metal charge transfer complexes as light absorbers, an integrated, inorganic device shows promise for applications in water splitting and CO2 sequestration. Through computational modelling, we aim to elucidate electronic processes which occur on the nanoscale, with the ultimate goal of improving efficiency and durability.
Recent articles |
Networks and self assembly
|
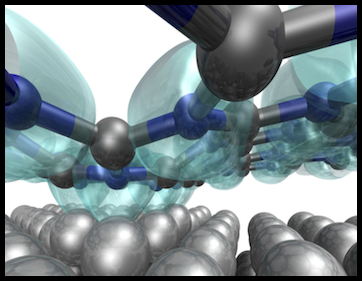
Organic-metallic interfaces offer the possibility of novel next-generation photovoltaic devices.
Producing these devices reliably and cheaply poses many challenges, however.
Understanding and controlling self-assembly processes is an important aspect of the realization of this technology. Interestingly, this work has recently spun off in a very different direction: Online social Networks. See our open source tool, #k@ (http://hashkat.org), for more information.
Recent articles |