|
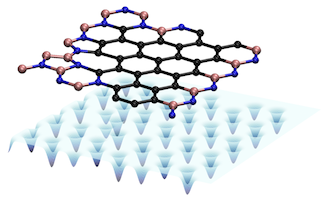
In this article, we demonstrate the ability of deep convolutional neural networks to learn the mapping between the atomic structure of a material to its total energy. We look at the particular case of 2-dimensional lattices made of boron, nitrogen, and carbon. The total energy we learn is computed using an approximate form of quantum mechanics known as density functional theory (techincally DFT isn't approximate, but the version we used was). The advantage of our approach is that it is able to make a calculation significantly faster (over 1000x) than is possible using DFT. This is a big deal, because DFT is a widely used computational tool in chemistry, physics, and material science. Althought our convolutional neural network is very accurate and efficient, we also demonstrate that it is not particularly good at extrapolating. If it sees a structure which is very different than what it trained on, it does not do a good job predicting the energy (or distance, in the case of two atom systems). We are currently working to improve this shortcoming.
|