|
Quantitative Molecular Orbital Energies within a G0W0 Approximation
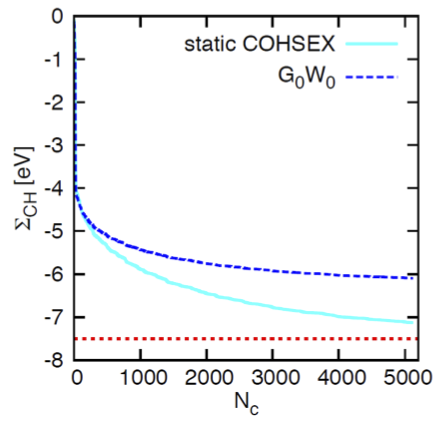
S. Sharifzadeh, I. Tamblyn, P. Doak, P. Darancet, and J.B. Neaton European Physical Journal B (2012) This work explores routes for computing the ionization potential, electron affinity, and fundamental gap of gas-phase molecules using many-body perturbation theory within the G0W0 approximation. The authors demonstrate that when employing an effective completion strategy for the unoccupied subspace and a properly converged dielectric function cutoff, the computed ionization potentials and electron affinities achieve excellent quantitative agreement with available experimental data, falling within 0.2 eV. The study establishes that a one-shot G0W0 approach is highly accurate for calculating addition and removal energies in small organic molecules, while identifying the dielectric function kinetic energy cutoff as a potentially limiting factor for broader application to larger organic systems. |