|
Theoretical design of redox levels of thiophene on functionalized light-absorbing semiconductor surfaces
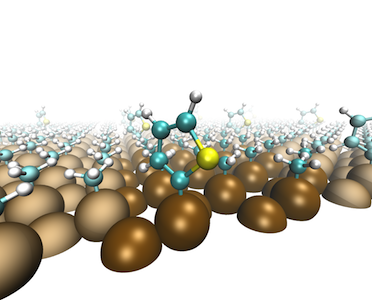
M. Yu, P. Doak, I. Tamblyn, and J.B. Neaton Journal of Physical Chemistry Letters, 4, 1701-1706 (2013) This work presents a general, empirical parameter-free approach for computing and understanding frontier orbital energies (redox levels) of covalently bonded organic-semiconductor interfaces. Using density functional theory and many-body perturbation theory within the GW approximation, the authors study thiophene molecules bonded to functionalized silicon (111) surfaces. The calculations reveal that standard DFT predictions require corrections of approximately 1 eV, with nonlocal electrostatic polarization effects contributing roughly 1.6 eV. Silicon band edge energies are predicted to vary by more than 2.5 eV with different functional groups, while molecular orbital energies remain relatively constant. These findings demonstrate the prospect of tuning energy alignment over a wide range through judicious selection of surface functional groups, with implications for photoelectrochemistry and related applications. |